
Background
Glycogen-storage disease type II (GSD II), also known as Pompe disease, is part of a group of metabolic diseases called lysosomal storage disorders (LSDs). GSD II is an autosomal-recessive disorder that results from deficiency of acid alpha-glucosidase (also known as acid maltase), a lysosomal hydrolase. The cellular role of acid alpha-glucosidase is to convert glycogen into glucose within the lysosomes. The Danish pathologist Joannes Cassianus Pompe first described this disease in 1932 when he was presented with a 7-month-old girl who died after developing idiopathic hypertrophic cardiomyopathy. Pompe observed an abnormal accumulation of glycogen in all postmortem tissues examined and described the cardinal pathologic features of this lysosomal storage disorder.
Pompe disease (GSD II) has a broad clinical spectrum. Classification is based on age of onset, organ involvement, severity, and the rate of disease progression. Three major forms of GSD II are recognized: classic infantile-onset, non-classic variant of infantile-onset, and late-onset (includes childhood, juvenile, and adult-onset).
Classic infantile-onset Pompe disease may be apparent in utero, but more often presents within the first two months of life. In this form, cardiac, skeletal, and respiratory muscles are involved. Clinical hallmarks of classic infantile-onset Pompe disease include hypotonia, generalized muscle weakness, cardiomegaly, hypertrophic cardiomyopathy, feeding difficulties, failure to thrive, respiratory distress, and hearing loss. Classic infantile-onset GSD II is marked by a progressive and rapidly fatal course. Without enzyme replacement therapy (ERT), classic infantile-onset Pompe disease commonly results in death within the first year of life due to cardiac disease from progressive left ventricular (LV) outflow obstruction.
The non-classic variant of infantile-onset Pompe disease presents within the first year of life. Patients older than 6 months present with motor delays and/or slowly progressive muscle weakness. Death results from ventilatory failure in early childhood. In this form, cardiac disease is not found to be a major cause of morbidity; however, cardiomegaly may be seen.
Late-onset GSD II is characterized by proximal muscle weakness and respiratory compromise. Adults with late-onset GSD II typically present with proximal muscle weakness between the second and sixth decades of life. These individuals ultimately die of respiratory failure. Cardiac involvement is less likely among individuals with disease onset at an older age.
The photomicrographs below document liver findings in GSD II.
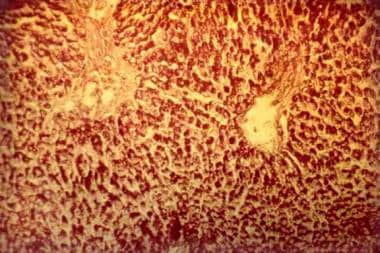
Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).

Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).