
Overview
von Hippel-Lindau (VHL) disease, or von Hippel-Lindau syndrome, is a rare genetic disorder characterized by visceral cysts and benign tumors in multiple organ systems that have subsequent potential for malignant change.
Clinical hallmarks of VHL disease include the development of retinal and central nervous system (CNS) hemangioblastomas (blood vessel tumors), pheochromocytomas, multiple cysts in the pancreas and kidneys, and an increased risk for malignant transformation of renal cysts into renal cell carcinoma. The wide age range and pleiotropic manner in which VHL disease presents complicates diagnosis and treatment in affected individuals, as well as their at-risk relatives. The image below illustrates a hemangioblastoma of the retina as found in patients with VHL disease.
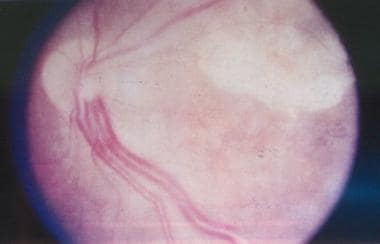
von Hippel-Lindau disease. Clinical picture of the retina, showing a pair of dilated and tortuous feeder vessels coursing on the surface of the retina from the optic nerve head toward the angioma (which is not seen in this image because it is in the extreme periphery).
Hemangiomas
VHL disease is characterized by retinal capillary hemangiomas (also called benign vascular hamartomas).
Diagnosed in 50% of patients with VHL disease, these hemangiomas are composed of endothelial cells and pericytes. The foamy stromal cells between the capillaries stain positive for glial fibrillary acid protein and neuron-specific enolase.
Retinal capillary hemangiomas, usually supplied by large dilated feeder vessels, may occur in any part of the retina. Serum leakage from these vessels and hemangiomas leads to retinal exudates. Organized fibroglial bands with traction retinal detachment and vitreous hemorrhage may occur, along with potential complications such as glaucoma or permanent vision loss.
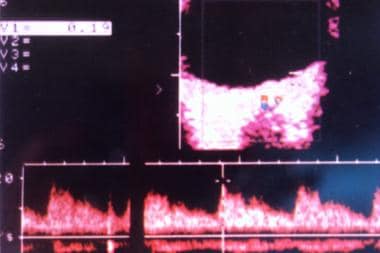
von Hippel-Lindau disease. Spectral display of the aberrant feeder vessels.
Tumors involving other organs and the CNS (brain, spinal cord) are present in 25% of patients with VHL disease.
Pancreatic and renal cysts can also occur and may be present concurrently.
Diagnostic considerations
The unexpected finding of a retinal or CNS hemangioblastoma or the diagnosis of a pheochromocytoma should prompt a search for other associated VHL disease features, as many of these patients may have the diagnostic criteria for VHL disease. Early identification of VHL is important because of the increased risk of serious complications (eg, renal cell carcinoma) to foster more effective treatment options and better prognoses.
Because VHL disease is a multiple-organ disease that widely varies in clinical presentation, various manifestations may lead to diagnosis. Criteria are the following:
More than one hemangioblastoma in the CNS (brain, spinal cord) or eye
A single hemangioblastoma in the CNS or retina, plus a visceral manifestation (multiple renal, pancreatic, or hepatic cysts; pheochromocytoma; renal cancer)
Positive family history plus any one of the above clinical manifestations
Elucidation of a deleterious mutation in the VHL gene
Genetic testing for mutations in the VHL gene is performed at many laboratories throughout the United States and the world. Gene Tests (www.genetests.org) cites 48 different laboratories in the United States that can test for the VHL gene mutation. Some of these locations are as follows:
Boston University School of Medicine, Center for Human Genetics, Boston, Massachusetts
Children’s Mercy Hospital, Molecular Genetics Laboratory, Kansas City, Missouri
Johns Hopkins Hospital, DNA Diagnostic Laboratory, Baltimore, Maryland
University of Pennsylvania School of Medicine, Genetic Diagnostic Laboratory, Philadelphia, Pennsylvania
Epidemiology
VHL disease is inherited in an autosomal-dominant Mendelian pattern. De novo mutations occur in about 1:4.4 million live births and account for 20% of cases. The incidence of VHL disease in the United States is approximately 1 case in 36,000 live births (worldwide incidence is 1:32,000 live births). Males and females are equally affected, and the diagnosis is made in people of all ethnic groups.
Age at diagnosis varies from infancy to age 60-70 years, with an average patient age at clinical diagnosis of 26 years. Clinically significant issues typically arise in affected individuals who are in their teens or twenties. About 20% of children with VHL can have ocular or adrenal signs before age 10 years. People can also first present with clinical findings in their ninth decade of life.