
Background
I-cell disease is an inherited lysosomal storage disorder.
It first was described in 1967 by Leroy and DeMars when they reported a patient with clinical and radiographic features similar to those of Hurler syndrome (mucopolysaccharidoses 1H [MPS 1H]) but with an earlier onset of symptoms and no evidence of mucopolysacchariduria.
One unique feature of this disease was the presence of phase-dense intracytoplasmic inclusions in the fibroblasts of patients. These cells were termed inclusion cells, or I-cells; thus, the disease was designated I-cell disease. Spranger and Wiedermann subsequently classified this disease as mucolipidosis type II (ML II) because it had clinical characteristics that included mucopolysaccharidoses and sphingolipidoses.
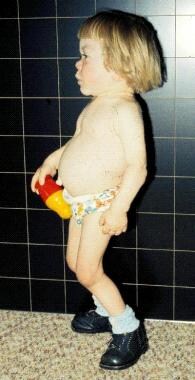
Profile view of 3-year-old with I-cell disease. Growth ceased more than one year earlier. Note small orbits, proptotic eyes, full and prominent mouth caused by gingival hypertrophy, short and broad hands, stiffening of small hand joints, prominent abdomen with umbilical hernia, and limited extension of the hips and knees.