
Practice Essentials
Lipodystrophy, the abnormal distribution of fat, encompasses lipoatrophy, the loss of fat, and other abnormalities of adipose tissue. Generalized lipoatrophy, also known as Lawrence-Seip syndrome, is a rare group of syndromes, either acquired or congenital, characterized by a near-complete absence of fat. See the image below.
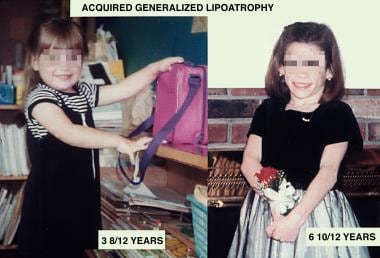
Acquired generalized lipoatrophy is in the differential diagnosis of congenital generalized lipoatrophy. This patient, who had completely normal fat distribution at age 3 years 8 months (left), acutely lost her body fat with a febrile illness at age 6 years 10 months (right).
Terminology
See the list below:
Lipoatrophy syndromes: Heterogeneous cluster of conditions in which the common trait is a lack of fat tissue, either generalized or partial and congenital or acquired
; the metabolic consequences include peripheral insulin resistance and hypertriglyceridemia
Lipodystrophy: Abnormal distribution of fat; consists of lipoatrophy and other abnormalities of adipose tissue
Lipoatrophy: Loss of fat
Focal lipoatrophy: Loss of fat involving a single body region, usually not associated with metabolic consequences (eg, gluteal area, usually following intramuscular injections
; face [Romberg syndrome]
; therapy with protein hormones such as insulin
and growth hormone
)
Lipoatrophic diabetes: Diabetes mellitus in association with lipoatrophy
Classification of lipoatrophy syndromes
See the list below:
Congenital generalized lipoatrophy (or, Seip-Berardinelli syndrome): Autosomal recessive; generalized absence of fat within the first year of life, followed by insulin resistance, acanthosis nigricans, diabetes mellitus before adolescence; severe hypertriglyceridemia accompanied by frequent pancreatitis; high basal metabolic rate
; increased appetite
Other genetic lipoatrophy syndromes: Autosomal dominant; characterized by an absence of fat from the face, upper trunk, arms, and below the knees; associated with a progressive neurodegenerative disorder (more severe in females) and congenital cataracts; insulin resistance; dyslipidemia (more severe in females)
Familial partial lipodystrophy (or, Kobberling-Dunnigan syndrome, familial partial lipodystrophy [FPLD] or Dunnigan syndrome): Autosomal dominant; affects primarily white persons of northern European descent but also some people of pedigrees of Asian descent; difficult to discern affected from unaffected patients before the onset of puberty; normal fat distribution at birth but a loss of subcutaneous fat in the extremities and/or trunk with early puberty, followed by increased fat in the face and neck with completion of puberty
; visceral fat and interfascicular intramuscular fat depots are preserved
Acquired lipoatrophy syndromes: Likely autoimmune in nature; panniculitis may precede fat loss in some cases
; severe fat loss leads to dramatic change in physical features (eg, loss of retro-orbital fat; loss of supportive fat of hands, feet, and genital area); increased basal metabolic rate and hepatic glucose output; diabetes
Other causes of acquired lipodystrophy: Aggressive treatment of patients infected with human immunodeficiency virus, particularly with protease inhibitors (partial lipoatrophy: loss of subcutaneous fat from the face, extremities, and trunk)
; increased visceral fat, with buffalo hump,
accompanied by insulin resistance and hypertriglyceridemia; affects children and adults of both sexes
Pathophysiology
Mechanisms for adipocyte deficiency include the following:
Defects in adipogenesis pathway
Increased destruction of adipocytes (eg, induction of apoptosis)
Potential mechanisms that may explain insulin resistance and other metabolic abnormalities caused by fat loss in lipoatrophy syndromes include the following
:
Inability to store triglycerides: Leads to abnormal deposition of triglycerides in other tissues (eg, liver, skeletal muscle, and pancreas [lipotoxicity]), resulting in hypertriglyceridemia, insulin resistance, hepatic steatosis (fatty liver), impaired insulin secretion, and, eventually, type 2 diabetes
Absent or immature adipocytes: Inability to synthesize and release adipocytokines (eg, leptin, adiponectin) that may be important to maintain normal metabolism
Clinical abnormalities in lipoatrophy/lipodystrophy syndromes
See the list below:
Insulin resistance and dyslipidemia
Increased circulating free fatty acids
Excessive accumulation of triglycerides in multiple sites, especially the liver and muscles
Elevated respiratory quotient
Management
The clinical problems requiring treatment in lipoatrophy syndromes are diabetes and hypertriglyceridemia.
Diabetes
Although several classes of antidiabetic drugs are marketed in the United States, achieving good glycemic control (glycated hemoglobin < 7.2%) is difficult for most patients with lipoatrophic diabetes.
Pharmacotherapy for lipoatrophy/lipodystrophy syndromes includes the following:
Leptin analogs (eg, metreleptin)
Insulin: Extremely high doses (eg, >1000 U/d) are often necessary in patients with various syndromes of lipoatrophy
Insulin secretagogues
Metformin
Acarbose
Thiazolidinediones
Hypertriglyceridemia
The dyslipidemia of severe lipoatrophy is difficult to treat with currently available therapeutic interventions. In addition, lifestyle modifications alone may not be sufficient and may need to be implemented along with pharmacotherapy.
Troglitazone is remarkable in its ability to improve hypertriglyceridemia in addition to improving glycemia. Some clinicians also prefer to use fibrates and statins rather than niacin because of severe insulin resistance.
In women with lipoatrophy syndromes, oral estrogens should be avoided as they can exacerbate the hypertriglyceridemia and result in acute pancreatitis.