
Practice Essentials
Regardless of the etiology, a deficiency of cyclooxygenase (COX), a key regulatory enzyme in the synthetic pathway of eicosanoid production, results in beneficial and detrimental physiologic conditions relative to imbalances of the eicosanoids.
Thus, tracing research of the COX pathway is essential to an understanding of COX deficiency, and examining the variable effects of COX inhibition are advantageous.
Eicosanoids, which include prostaglandins, leukotrienes, thromboxanes, and lipoxins, are responsible for multiple inflammatory, mitogenic, and angiogenic activities in various tissue and organ systems. Therefore, COX — also known as prostaglandin-endoperoxide synthase (PTGS), fatty acid COX, prostaglandin H (PGH) synthase, and EC 1.14.99.1 — is implicated in the production of fever, inflammation, and pain.
In 1930, American gynecologists Kurzok and Lieb first described the stimulatory effects of seminal fluid on human uterine muscle tissue. A few years later, von Euler of Sweden independently discovered similar effects of human seminal fluid on smooth muscle tissue. He localized the biologic activity to a fraction of lipid soluble acids that he termed “prostaglandin,” hypothesizing that these substances originate in the prostate gland. Two decades later, the prostaglandins were deduced to be a family of related compounds that contain 20-carbon polyunsaturated fatty acids with a cyclopentane ring, as depicted below.
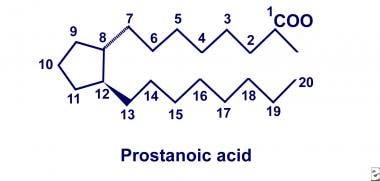
Twenty-carbon polyunsaturated fatty acid with cyclopentane ring.
By 1964, after recognition of this basic structure, Bergstrom and colleagues successfully synthesized series 2 prostaglandins from arachidonic acid using sheep seminal fluid. However, the physiologic significance of prostaglandin production did not unfold until 1971, when Vane, Smith, and Willis discovered that aspirin and indomethacin inhibited prostaglandin biosynthesis. Further investigations by Smith concluded that aspirin and indomethacin inhibited synthesis by specifically blocking oxygenation of arachidonic acid. Collectively, these landmark discoveries provided initial insight into the COX pathway of arachidonate metabolism.
Advances in genomic analysis have led to a clearer understanding of the COX pathway. Initial investigations by Miyamoto and Simmons demonstrated that 2 isoforms exist: COX-1 (PTGS-1) and COX-2 (PTGS-2), respectively. The transcription of COX-1 yields a 2.7-kilobase (kb) messenger ribonucleic acid (mRNA) that encodes a 576-residue, 65-kd protein. Conversely, the transcription of COX-2 yields a 4.5-kb mRNA that encodes a 70-kd protein with roughly 70-75% homology to the COX-1 protein.
Funk and co-investigators localized COX-1 to 9q32-q33.3 via somatic hybrid deoxyribonucleic acid (DNA) analysis. Tay and colleagues then localized COX-2 to 1q25.2-q25.3 via fluorescein in situ hybridization. Furthermore, using sequence analysis of human genomic DNA, researchers concluded that the amino acids important for catalysis by COX-1 are conserved and are equally important for catalysis by COX-2.
Activity of COX-1 and COX-2
Evidence suggests that COX-1 and COX-2 are similar in structure and function but that they exist as 2 distinct enzymatic entities. They have been defined as monotropic integral membrane proteins located primarily in the endoplasmic reticulum (COX-1) and the perinuclear envelope (COX-2). Their distinct biosynthetic activity includes an endoperoxidase synthase reaction that oxygenates and cyclizes polyunsaturated fatty acid precursors (eg, arachidonic acid) to form prostaglandin G2 (PGG2), and a peroxidase reaction that converts PGG2 to prostaglandin H2 (PGH2), as shown below. In turn, PGH2 is converted to biologically active products (ie, prostaglandin E2 [PGE2]) by individual synthase and reductase reactions.
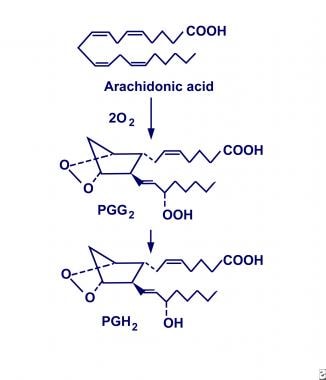
Cyclooxygenase conversion of arachidonic acid into prostaglandin H2 (PGH2).
COX-1 is expressed constitutively and is isolated throughout most cell lines in almost all mammalian tissues. It is described as a housekeeping enzyme, being responsible for cell-to-cell signaling, tissue homeostasis, and cytoprotection. In view of this, researchers hypothesize that COX-1 is a rapid responder to various physiologic conditions.
Conversely, COX-2 is described as an inducible isoform influenced by a plethora of proinflammatory mediators. Without appropriate stimulation, isolation of the COX-2 protein is negligible in most tissues. However, newer literature reveals that COX-2 is expressed constitutively in some cell lines of the brain, kidney, and trachea. Although vaguely described, COX-2 is considered to be a principal mediator of inflammation, mitogenesis, and angiogenesis.
Studies have demonstrated that the eicosanoids produced by cytosolic COX-1 participate in autocrine and paracrine activities, while those produced by perinuclear COX-2 result in intracrine activity. This subcellular localization of the COX enzyme (through the use of new microscopy techniques) has helped to explain why 2 isoforms exist. Furthermore, researchers have proposed that COX-1 and COX-2 acquire arachidonic acid from different phospholipases, suggesting participation through separate pathways.
Etiology
COX deficiency typically denotes an acquired cause-and-effect relationship between the protein’s enzymatic activity and nonsteroidal anti-inflammatory drugs (NSAIDs). However, Nyman and co-investigators, while studying bleeder families on the Aland Islands, reported an autosomal dominant pattern of inheritance for COX deficiency.
In addition, Horellou and colleagues described 3 family members of 2 successive generations with bleeding tendencies and concluded that their platelets were compatible with a COX deficiency.
This study also described an autosomal dominant pattern of inheritance but deduced that COX was not imperative to aggregation and adenosine triphosphate (ATP) release in the face of high collagen concentrations. Sporadically documented familial and constitutional bleeding disorders have been associated with platelet COX-1 deficiency.