
Practice Essentials
A glycogen storage disease (GSD) is the result of enzyme defects in the glycogen pathway. These enzymes normally catalyze reactions that ultimately convert glycogen compounds to glucose. Enzyme deficiency results in glycogen accumulation in tissues.
In many cases, the defect has systemic consequences, but in some cases, the defect is limited to specific tissues. Most patients experience muscle symptoms, such as weakness and cramps, although certain GSDs manifest as specific syndromes, such as hypoglycemic seizures or cardiomegaly.
GSD VI is caused by deficient activity of hepatic glycogen phosphorylase, an enzyme encoded by the PYGL gene, which is located on chromosome 14q21-q22. PYGL is the only gene known to be associated with GSD VI.
With an enzyme defect, carbohydrate metabolic pathways are blocked and excess glycogen accumulates in affected tissues. Each GSD represents a specific enzyme defect, and each enzyme is either focal or systemic. Hepatic phosphorylase, which is found in the liver and red blood cells, is deficient in GSD VI, which results in glycogen accumulation in the liver and subsequent hypoglycemia. These inherited enzyme defects usually present in childhood, although some, such as McArdle disease and Pompe disease (also known as acid maltase deficiency), have separate adult-onset forms. In general, GSDs are inherited as autosomal recessive conditions. Several different mutations have been reported for each disorder.
Diagnosis depends on findings from patient history and physical examination, muscle biopsy, electromyography, ischemic forearm testing, and creatine kinase testing. Biochemical assay for enzyme activity is the method of definitive diagnosis.
Individuals with GSD VI can present with hepatomegaly with elevated serum transaminases, ketotic hypoglycemia, hyperlipidemia, and impaired growth. Symptoms result from mild hypoglycemia. Liver fibrosis and hepatocellular carcinoma have been reported in patients with GSD VI.
A creatine kinase level evaluation is helpful in all cases of suspected glycogen storage disease (GSD). Because hypoglycemia may be found in some types of GSD, fasting glucose testing is indicated. In Hers disease, hypoglycemia is a primary concern. Urine studies are indicated because myoglobinuria may occur in some patients with GSDs. Hepatic failure occurs in some patients with GSDs, although rarely in those with Hers disease. Liver function studies are indicated and may reveal evidence of hepatic injury. Biochemical assay of enzyme activity is necessary for definitive diagnosis. Findings from imaging studies may reveal hepatomegaly.
Liver biopsy may be required to diagnose the cause of hepatomegaly. Identification of 2 pathogenic variants in trans in PYGL confirms a diagnosis of GSD VI. About 30 pathogenic variants have been reported throughout the PYGL gene.
The following list contains a quick reference for 8 of the GSD types:
0 – Glycogen synthase deficiency
Ia –Glucose-6-phosphatase deficiency (von Gierke disease)
II –Acid maltase deficiency (Pompe disease)
III – Debranching enzyme deficiency (Forbes-Cori disease)
IV – Transglucosidase deficiency (Andersen disease, amylopectinosis)
V – Myophosphorylase deficiency (McArdle disease)
VI – Phosphorylase deficiency (Hers disease)
VII – Phosphofructokinase deficiency (Tauri disease)
The chart below demonstrates where various forms of GSD affect metabolic carbohydrate pathways.
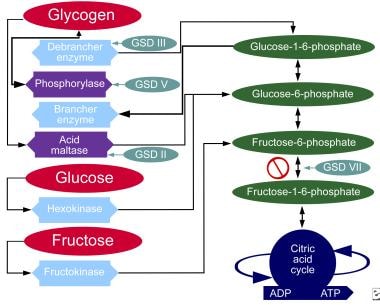
Metabolic pathways of carbohydrates.
Although at least 14 unique GSDs are discussed in the literature, the GSDs that cause clinically significant muscle weakness are Pompe disease (GSD type II, acid maltase deficiency)
, Cori disease (GSD type III, debranching enzyme deficiency), McArdle disease (GSD type V, myophosphorylase deficiency), and Tarui disease (GSD type VII, phosphofructokinase deficiency). One form, von Gierke disease (GSD type Ia, glucose-6-phosphatase deficiency), causes clinically significant end-organ disease with significant morbidity. Interestingly, GSD type 0 also is described, which is due to defective glycogen synthase.