
Practice Essentials
Dystrophin protein is integral to the structural stability of the myofiber. Without dystrophin, muscles are susceptible to mechanical injury and undergo repeated cycles of necrosis and regeneration. Duchenne and Becker muscular dystrophies are caused by mutations in the same gene encoding dystrophin. These disorders almost exclusively affect males because of the X-linked inheritance pattern. See the image below.
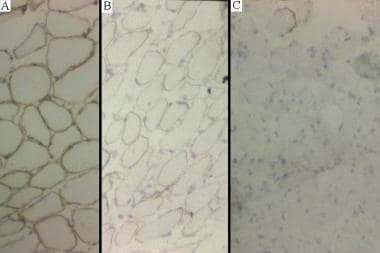
(A) Normal dystrophin staining.(B) Intermediate dystrophin staining in a patient with Becker muscular dystrophy.(C) Absent dystrophin staining in a patient with Duchenne muscular dystrophy.
Signs and symptoms
Diagnostic criteria include the following:
Weakness with onset in the legs
Hyperlordosis with wide-based gait
Hypertrophy of weak muscles
Progressive course over time
Reduced muscle contractility on electrical stimulation in advanced stages of the disease
Absence of bladder or bowel dysfunction, sensory disturbance, or febrile illness
Stage 1 – Presymptomatic
Progression of muscular dystrophy occurs in 5 stages. In stage 1, creatine kinase levels are usually elevated. Patients have a positive family history.
Stage 2 – Early ambulatory
Waddling gait, manifesting in children aged 2-6 years; often the first clinical symptom in patients with Duchenne muscular dystrophy and is secondary to hip girdle muscle weakness
Inexorable progressive weakness in the proximal musculature, initially in the lower extremities, but later involving the neck flexors, shoulders, and arms
Because of proximal lower back and extremity weakness, parents often note that the boy pushes on his knees in order to stand; this is known as the Gowers sign
Possible toe-walking
Can climb stairs
Stage 3 – Late ambulatory
More difficulty walking
Around age 8 years, most patients notice difficulty with ascending stairs, and respiratory muscle strength begins a slow but steady decline
Cannot arise from the floor
At approximately the same time as independent ambulation is most challenged, the forced vital capacity begins to gradually wane, leading to symptoms of nocturnal hypoxemia such as lethargy and early morning headaches
Stage 4 – Early nonambulatory
Can self-propel for some time
Able to maintain posture
Possible development of scoliosis
Stage 5 – Late nonambulatory
Scoliosis may progress, especially when more wheelchair dependent
If wheelchair bound and profoundly weak, patients develop terminal respiratory or cardiac failure, usually by the early 20s, if not sooner; poor nutritional intake can also be a serious complication in individuals with severe end-stage Duchenne muscular dystrophy
Contractures may develop
See Clinical Presentation for more detail.
Diagnosis
Serum creatine phosphokinase (CPK) level
Always increased in patients with Duchenne muscular dystrophy or Becker muscular dystrophy, probably from birth
Often increased to 50-100 times the reference range (ie, as high as 20,000 mU/mL)
Decreases over time; in late-stage DMD, very little muscle mass remains to give rise to an elevated serum CPK level
Strongly suspect Duchenne muscular dystrophy in a child with proximal weakness and very elevated levels of CPK
Imaging studies
Radiographs of the spine are important for screening and evaluating the degree of scoliotic deformity
Chest radiography is often part of the evaluation as the disease progresses and dyspnea develops
Beyond imaging for scoliosis and dyspnea, imaging studies are of little help in making the diagnosis
Dual energy x-ray absorptiometry estimates bone mineral density, as individuals with dystrophinopathies can have accelerated osteopenia/osteoporosis/fracture risk
Electromyography
Electromyography (EMG), even though not diagnostic, narrows the differential diagnosis by effectively excluding primarily neurogenic processes such as spinal muscular atrophy
In general, the proximal muscles of the lower extremities may exhibit the more prominent EMG findings
A sufficient number of muscles must be sampled to establish the presence of a diffuse process such as a dystrophy
Molecular diagnosis
Duchenne or Becker muscular dystrophy can be reliably and accurately detected from peripheral blood samples in nearly all cases
If deletion/duplication genetic tests are uninformative, direct sequencing of the dystrophin gene is a viable option
Muscle biopsy
Required for diagnosis in patients without detectable mutations of the dystrophin gene
For some families of a young boy found to have a dystrophin mutation, the muscle biopsy can provide critically important dystrophin protein information such as molecular weight size and abundance
Immunolabeling of frozen muscle sections can enable epitope identification
Depending on the purpose of the biopsy, proper site selection is crucial
Cardiac assessment
ECG can uncover sinus arrhythmias and may show deep Q waves and elevated right precordial R waves
Transthoracic echocardiography often reveals small ventricles with prolonged diastolic relaxation
A Holter monitor is valuable for paroxysmal arrhythmias
Cardiac MRI and gadolinium enhancement can better characterize cardiac tissue changes
See Workup for more detail.
Management
Therapeutic strategies for the dystrophinopathies can be categorized into the following 3 groups:
Supportive pharmacologic therapy
Research gene therapy
Research cellular therapy
No cure yet exists for Duchenne or Becker muscular dystrophy, but medical and supportive treatments can reduce morbidity, improve quality of life, and prolong lifespan.
Supportive therapy
Corticosteroids are the only proven medical treatment for Duchenne muscular dystrophy
Benefits include prolongation of ambulation, maintenance of strength and function, and delay in the development of scoliosis
Clinical improvement is seen as early as 1 month after starting treatment and can last as long as 3 years
Controversies exist with respect to the age at which to start corticosteroids, clinical criteria for starting corticosteroids, which corticosteroid to use, which dose and which regimen (continuous daily or intermittent) to use, and when to discontinue corticosteroid
ACE inhibitors may provide benefit in patients with and without ventricular dysfunction
With the supervision of an experienced physical therapist, daily joint-stretching exercises is important for parents to incorporate into the home regimen
Night splints can have a favorable influence
Judicious use of tendon release surgeries may prolong ambulation by as long as 2 years
Braces can be therapeutic adjuncts, but currently are less often used
Gentle sports or activities (eg, swimming, tricycle/bicycles) may be encouraged
TREAT-NMD has published standards of care for Duchenne muscular dystrophy.
See Treatment and Medication for more detail.