
Overview
Due to the efforts of numerous clinicians and basic scientists over many years, we now have a firm understanding of how the motor signal generated in the brain travels down the spinal cord, into a peripheral nerve, and interfaces with the target muscle. Furthermore, we are now able to analyze many aspects of this complex process in the clinic or at the bedside via clinical examination findings, pharmacological challenge, or electrophysiological equipment (see the image below). These advances in scientific understanding that have directly impacted clinical efficacy represent an excellent example of translational research.
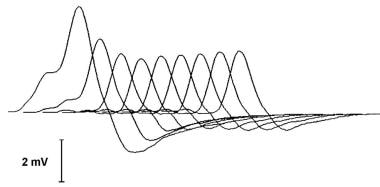
A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
When discussing disease of the neuromuscular junction (NMJ), many physicians first think of myasthenia gravis (MG). This is entirely appropriate, as research into the pathogenesis of MG has always been at the center of our understanding of the NMJ. The first patient with MG described in medical writings was likely a Native American named Chief Opechankanough, uncle of the famous Matoaka (Pocahontas). Colonial physicians who examined him in the early 1600s documented weakness and ptosis that improved with rest. Their accounts were not formally published in a medical journal.
However, in 1672, Thomas Willis (for whom the Circle of Willis is named) published the case of an English woman whose clinical findings seemed consistent with MG.
Numerous cases were subsequently described. Several accounts were documented by William Erb in 1879, and in 1893, Samuel Goldflam provided excellent clinical descriptions. Friedrich Jolly coined the term myasthenia gravis pseudoparalytica in 1895 and also pioneered one of the assessment methods, repetitive stimulation, which is discussed later.
The next advance occurred in the 1930s when English doctor Mary Broadfoot Walker, one of the first female British physicians, showed that injections of physostigmine could temporarily increase the strength of individuals with MG and reverse symptoms such as ptosis. She reported these findings in Lancet in 1934. The Tensilon test, discussed later, is the modern version of Dr. Walker’s technique. Her work, particularly when combined with the basic pharmacologic studies of contemporaries such as Dale and Feldberg on cholinergic neuromuscular transmission, suggested impairment of cholinergic transmission at the nerve-muscle synapse.
Approximately 30 years prior to Dr. Walker’s findings, more insight into to pathophysiology of MG had emerged. In 1901, the famous pathologist Karl Weigert noted that lymphoid cells were present in the muscle and other tissues of persons with this affliction. He also made the connection between MG and thymic hypertrophy and/or neoplasia. Four years later, neurologist E. Farquhar Buzzard noted collections of lymphocytes (lymphorrhages) in the tissues of patients with MG. Shortly after the observations of Weigert and Buzzard became known, clinicians began to use thymectomy as a treatment for MG.
Initially, only actual thymomas were resected, but the practice soon expanded to include thymic hyperplasia and ultimately removal of the thymus for patients without any apparent thymic abnormality.
Throughout the first part of the twentieth century, evidence for the immunological basis of MG continued to accumulate. In 1960, Dr John A. Simpson introduced his hypothesis that the cause of MG was “an ‘autoimmune’ response of muscle in which an antibody to end-plate protein may be formed.”
Nonetheless, many competing hypotheses remained active for the next 15 years. This was largely due to 2 primary points of contention: (1) Which side of the nerve-muscle junction was the defect in MG (the presynaptic nerve endings or the postsynaptic motor end plate)? and (2) What was the fundamental etiology of the disease (eg, autoimmune disease as Simpson had speculated)?
In the 1970s, new techniques emerged that allowed for further investigation of the function of the neuromuscular junction. In particular, the use of alpha-bungarotoxin, a compound derived from snake venom, that bound to specific portions of the acetylcholine receptor (AChR) was pioneered by pharmacologists C. C. Chang, C. Y. Lee, and L. F. Tseng. Further contributions from clinician-scientists such as Andrew Engel, Douglas Fambrough, Edward Lambert, Vanda Lennon, Daniel Drachman, Eric Stalberg, Joze Trontelj, Jan Ekstedt, John Newsom-Davis, David Richman, Marjorie Seybold, and Klaus Toyka helped to integrate the emerging immunologic-neurophysiologic connections suggesting that the disorder was secondary to immunologic dysfunction at the postsynaptic motor end plate.
Jim Patrick, Jon Lindstrom, and associates at the Salk Institute produced antibodies to various subunits of the AChR and ultimately demonstrated that the disease could be reproduced by antibodies to this receptor.
The highly detailed knowledge of neuromuscular transmission that we appreciate today is largely due to the efforts toward understanding MG and other diseases of the NMJ. To be certain, some aspects remain unknown. However, the essential framework is now common knowledge to most students early in medical school and can be summarized as follows:
A nerve action potential arrives at the axon terminal.
Voltage-gated calcium channels open and calcium ions enter the neuron.
A biochemical cascade ensues, causing vesicles containing acetylcholine (ACh) to fuse with the cell membrane, releasing ACh into the synaptic cleft.
ACh diffuses across the synaptic cleft and binds to nicotinic AChRs on the postsynaptic cell membrane.
The AChRs, which are sodium ion channels, open and allow sodium ions to flow into the muscle cell. Several other nearby membrane proteins including agrin, rapsyn, and muscle-specific tyrosine kinase (MuSK) influence both the function and the positioning of the AChRs.
The influx of sodium (accompanied by an exit of potassium through potassium channels) causes depolarization of the muscle cell membrane.
If depolarization is sufficient, it triggers a regenerative muscle action potential that spreads throughout the cell into the transverse tubules. This causes a release of calcium from the sarcoplasmic reticulum, which initiates muscle contraction.
When the ACh molecule is released from the AChR, it is degraded by the enzyme acetylcholinesterase (AChE) into acetate and choline. After reuptake by the neuron, choline is combined with another acetate group to reform acetylcholine, which is subsequently repackaged into a vesicle.
Our current understanding of neuromuscular transmission is now complemented by modern methods for diagnosis of dysfunction at the myoneural synapse. The primary components for clinical assessment are history and physical examination, pharmacologic challenge, neurophysiologic (electrical) studies and neuroimmunologic assays. Each of these are discussed in the following sections.