
Coagulation Disorders
Antiphospholipid syndrome
Antiphospholipid syndrome (APS) is an acquired, multisystemic disorder characterized by recurrent thromboses in the arterial system, venous system, or both. Antiphospholipid syndrome is classified into 2 groups: primary and secondary.
Secondary antiphospholipid syndrome is often associated with systemic lupus erythematous (SLE) and infrequently with other diseases, such as lymphoproliferative disorders, autoimmune diseases, infections (eg, syphilis, HIV, hepatitis C), and drugs (eg, procainamide, quinidine, hydralazine, phenytoin, chlorpromazine). Serologic markers for antiphospholipid syndrome are antiphospholipid antibodies (beta2-GPI or anticardiolipins) or lupus anticoagulant.
The APS diagnosis should meet the revised Sapporo criteria (also called the Sydney criteria) for definite APS. The criteria, developed by consensus among international clinical and laboratory experts, were originally referred to in 1999 as the Sapporo criteria
and then modified at a meeting in Sydney in 2006.
The primary diagnostic criteria include arterial thrombosis, venous thrombosis, recurrent fetal loss, and thrombocytopenia. One of the listed primary criteria is required for diagnosis, combined with a sustained elevated titer of immunoglobulin G (IgG) anticardiolipin or lupus anticoagulant, which can be detected based on the prolonged activated partial thromboplastin time (aPTT), Kaolin clotting time, or dilute Russell Viper venom time.
In a large retrospective study from the Mayo Clinic, 41% of patients with lupus anticoagulant had skin lesions as the first sign of antiphospholipid syndrome.
Cutaneous manifestations include livedo reticularis, necrotizing vasculitis, livedo vasculitis, thrombophlebitis, cutaneous ulceration and necrosis, erythematous macules, purpura, ecchymoses, painful skin nodules, and subungual splinter hemorrhages.
Livedo reticularis is a presenting sign in up to 40% of patients with the diagnosis of SLE.
Skin changes defined as livedo reticularis are violaceous, red or blue, reticular, or mottled pattern of the skin of the arms, legs, and the trunk. They are not reversible with rewarming.
Noninflammatory vascular thrombosis is the most frequent finding in skin lesions of patients with antiphospholipid syndrome. Differential diagnoses include cryoglobulinemia, warfarin-induced necrosis, purpura fulminans, emboli to the skin, thrombocythemia, protein C deficiency, Sneddon syndrome, and skin ulcers in patients with sickle cell anemia or hemolytic anemia. In some antiphospholipid syndrome lesions, hemosiderin deposition can make differentiation from Kaposi sarcoma difficult.
The mainstays of prophylaxis and treatment of thrombosis in patients with antiphospholipid syndrome are anticoagulant and antiplatelet agents. Immunosuppression and immunotherapy (eg, corticosteroids, pulse cyclophosphamide, plasmapheresis, gamma-globulin infusions) only transiently reduce elevated antibody levels. Indications for immunotherapy are limited to systemic lupus erythematous or catastrophic vascular occlusion syndrome.
No studies are available to show the usefulness of prophylaxis in patients with antibodies in the absence of thrombosis. Some authors suggest using low-dose aspirin in patients with persistent lupus anticoagulant or high levels of IgG anticardiolipin. Some patients with antiphospholipid syndrome may have resistance to a usual dose of subcutaneous heparin. Low molecular weight heparins may prove beneficial because of improved safety and efficacy profiles (eg, decreased bleeding, risk of osteoporosis and thrombocytopenia, prolonged half-life, increased bioavailability). The protective effect of hydroxychloroquine against thrombosis has been reported in patients with systemic lupus erythematous.
Regarding treatment, a lack of consensus exists concerning the intensity and the duration of anticoagulation for patients with antiphospholipid-antibody–associated thrombosis. Warfarin is the treatment of choice for patients with antiphospholipid syndrome. Moderate-intensity anticoagulation with warfarin (to an international normalized ratio [INR] of 2.0-3.0) with or without low-dose aspirin of 75 mg/d was as effective as high-intensity warfarin (to INR 3.0-4.0) in a randomized trial.
High-intensity warfarin therapy should be considered only in patients receiving moderate-intensity warfarin with recurrent thrombosis.
Patients with catastrophic antiphospholipid syndrome usually receive a combination of anticoagulants, corticosteroids, intravenous immunoglobulin (IVIG), and plasma exchange; however, despite this aggressive approach, the mortality remains high.
The Antiphospholipid Syndrome Alliance for Clinical Trials and International Networking, formed in 2010, concerns large-scale, multicenter clinical trials and research in persistently antiphospholipid antibody‒positive patients.
The 14th International Congress on antiphospholipid antibodies held in Rio de Janeiro in September 2013 systematically reviewed the new trends in treatment strategies, including new oral anticoagulants, hydroxychloroquine, statins, rituximab, and other B-cell–targeting agents.
Warfarin-induced skin necrosis
Warfarin-induced skin necrosis is an uncommon serious complication of oral anticoagulation therapy (see the image below).
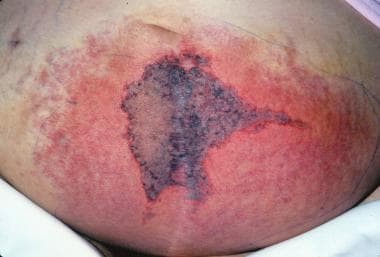
Warfarin (Coumadin)–induced skin necrosis on the lower abdomen.
Warfarin-induced skin necrosis is estimated to occur in 0.01-0.1% of patients treated with anticoagulants. Approximately 85% of reported patients are women. The most commonly affected sites are the breasts, buttocks, thighs, and abdomen, possibly because of the abundance of small dermal blood vessels in fatty tissue. Less usual locations include the external ear skin,
eyelids,
penis,
or digits.
A classic pattern of skin changes occurs when the condition commences. The initial nonspecific symptom may be a sensation of pain or cold in the affected area, followed by the development of a well-demarcated erythematous lesion that progresses to bullae formation and full-thickness skin necrosis. Areas of erythematous flush may become edematous and have a peau d’orange effect. The eschar may eventually slough or require extensive surgical debridement.
Most cases of warfarin-induced skin necrosis occur within the first week of warfarin administration, although rare cases of late onset are reported.
Biopsy specimens of lesions reveal involvement of the dermis and the subcutaneous tissue characterized by fibrin deposition in the small veins and postcapillary venules, with hemorrhage and diffuse necrosis in the dermis and the subcutaneous fat. Absence of arterial thrombosis is another characteristic feature.
Differential diagnoses include necrotizing fasciitis, venous gangrene, pyoderma gangrenosum, and cholesterol embolization syndrome. Warfarin is believed to induce a relative hypercoagulable state by interfering with the natural anticoagulant pathways (eg, those involving protein C, protein S, and antithrombin). A substantial minority of cases occurs in association with a familial deficiency of protein C or protein S. An acquired deficiency of protein S secondary to the development of antiphospholipid antibodies has been implicated. There is increasing recognition of the fact that skin necrosis could be the first presentation of heparin-induced thrombocytopenia.
Patients receiving large, rapidly administered loading doses of warfarin are at particular risk. Identifying patients at risk (eg, patients with a previous episode, protein C or protein S deficiency, or antiphospholipid syndrome), avoiding excessive initial doses of warfarin, and administering vitamin K in the early stages of lesion development can prevent warfarin-induced skin necrosis. If lesions have already progressed, oral anticoagulation agents should be discontinued. In some patients, attempts have been made to reverse the anticoagulation with plasma, and, in patients with documented protein C deficiency, concentrates of protein C have been used. Anticoagulation with heparin should be continued until the necrotic lesions heal. Despite treatment, approximately 50% of patients ultimately require skin grafting.
Warfarin also has been implicated in the etiology of calciphylaxis in patients with end-stage renal disease. It is a poorly understood disorder associated with high morbidity and mortality (60-80%). Clinically, patients present with painful, necrotic skin ulcers. Histopathology evaluation reveals medial calcification and intimal hyperplasia of small and medium-sized arteries of dermal and subcutaneous tissues.
The differential diagnosis includes vasculitis, cholesterol embolization syndrome, warfarin-induced skin necrosis, nephrogenic fibrosing dermopathy, ecthyma, cryofibrinogenemia, cellulitis, necrotizing fasciitis, coagulation disorders, and adverse drug effects. Treatment options are limited.